Adipose tissue aging mechanisms and systemic metabolic consequences
Adipose tissue is no longer viewed merely as an inert reservoir for organismal lipid storage. Contemporary endocrinology and geroscience have firmly established adipose tissue as a highly dynamic, central endocrine organ that dictates systemic energy homeostasis, immune regulation, and ultimately, organismal longevity. As chronological age advances, the adipose organ undergoes profound morphological, cellular, and molecular remodeling. Historically, scientific consensus and early epidemiological models portrayed all adiposity as uniformly detrimental to metabolic health. However, recent paradigms, heavily informed by single-cell transcriptomics and spatial omics of the 2020s, emphasize the critical, protective role of healthy subcutaneous adipose tissue (SAT). SAT serves as a safe metabolic sink, mitigating systemic lipotoxicity and preserving peripheral insulin sensitivity through the continuous secretion of beneficial adipokines 12. Conversely, the age-related decline in SAT expandability and adipogenic capacity forces a pathological redistribution of ectopic lipid into visceral adipose tissue (VAT) and non-adipose organs such as the liver and skeletal muscle, driving a systemic cascade of metabolic dysfunction 345.
Understanding the trajectory of adipose aging requires a comprehensive, multidimensional analysis. This encompasses the deterioration of the cellular microenvironment - specifically mitochondrial dysfunction, altered lipid droplet dynamics, and extracellular matrix (ECM) remodeling - as well as the immunometabolic shifts that transform the tissue into a primary source of chronic, low-grade systemic inflammation, colloquially termed "inflammaging" 67. Furthermore, ethnic and genetic variances significantly dictate visceral adiposity thresholds and metabolic risk trajectories across diverse global cohorts, demanding a shift away from universal anthropometric guidelines. Consequently, therapeutic interventions targeting adipose aging, including senolytics, incretin-based therapies (GLP-1/GIP agonists), and nutrient-sensing modulators (metformin, rapamycin), have entered intense clinical investigation. While these modalities hold immense promise, their clinical translation is highly debated, bounded by limitations such as off-target effects, senescent cell resistance mechanisms, and the critical risk of exacerbating age-related sarcopenia 891011.
The Protective Role of Subcutaneous Adipose Tissue and Mechanisms of Atrophy
To understand the pathology of the aging adipose organ, one must first recognize the physiological necessity of healthy subcutaneous adipose tissue. Developing perinatally, SAT is developmentally and functionally distinct from VAT, which primarily forms postnatally 2. In youthful, metabolically healthy individuals, SAT acts as the primary buffer against nutrient excess. By efficiently sequestering free fatty acids into inert triglycerides, healthy SAT prevents ectopic lipid deposition in the vasculature, myocardium, and hepatic parenchyma 28. The protective nature of SAT is particularly evident in premenopausal women, whose lipid distribution is predominantly localized to the gluteal-femoral subcutaneous compartment. This localized storage is heavily mediated by estrogen receptor alpha (ERα) expression, which drives lipoprotein lipase activity and facilitates safe triacylglycerol accumulation, thereby conferring significant protection against cardiovascular disease 3.
However, advancing age precipitates a relentless decline in SAT volume and functionality, a phenomenon closely linked to the exhaustion of adipocyte progenitor populations. Recent single-cell RNA sequencing (scRNA-seq) analyses have identified a unique, aging-dependent population of cells within the subcutaneous depots of both aged mice and humans. These Aging-dependent Regulatory Cells (ARCs) are of the fibroblast lineage and express traditional adipose progenitor markers, but completely lack adipogenic capacity 2. Driven by the transcription factor PU.1, ARCs actively secrete high levels of pro-inflammatory chemokines, such as CCL6, which autonomously inhibit the proliferation and differentiation of neighboring healthy adipose precursors 2.
As the subcutaneous depot loses its ability to generate new, small, insulin-sensitive adipocytes (hyperplasia), existing mature adipocytes are forced to undergo extreme hypertrophy to handle incoming lipid loads. These massively enlarged cells suffer from altered lipid droplet membrane architecture, severe mechanical stress, and localized hypoxia. The resulting failure of the subcutaneous depot to efficiently sequester lipids forces fatty acid spillover into the systemic circulation. This systemic lipotoxicity is the primary pathophysiological driver for the age-related expansion of visceral fat, tightly linking subcutaneous adipogenic failure to the onset of systemic insulin resistance and metabolic syndrome 249. In postmenopausal women, the decline in estrogen and the subsequent rise in the androgen-to-estrogen ratio drastically accelerate this lipid redistribution from the relatively inert gynoid stores to the highly metabolically active and pathogenic abdominal visceral compartment 31011.
Cellular and Molecular Hallmarks of Adipose Aging
The systemic failure of adipose tissue originates from intrinsic cellular deterioration. As the organ ages, the highly coordinated processes of mitochondrial respiration, lipid droplet dynamics, and structural tissue remodeling begin to fail, fundamentally altering the microenvironment and establishing a localized state of cellular distress.
Mitochondrial Dysfunction and Thermogenic Decline
Mitochondrial integrity is paramount for adipocyte function, governing not only fundamental ATP production but also lipogenesis, fatty acid oxidation, and the regulation of reactive oxygen species (ROS). During aging, adipose tissue exhibits a marked accumulation of mitochondrial DNA (mtDNA) damage, reduced mitochondrial biogenesis, and a general decline in mitochondrial respiratory capacity 45. This dysfunction manifests through impaired mitochondrial dynamics - specifically a disruption in the balance between mitochondrial fission and fusion - alongside defective mitophagy. The failure to clear damaged mitochondria results in the leakage of mtDNA into the cytosol. This cytosolic mtDNA acts as a highly potent damage-associated molecular pattern (DAMP), triggering the cGAS-STING innate immune pathway and exacerbating local tissue inflammation 1612.
Furthermore, mitochondrial dysfunction critically impairs the thermogenic capacity of both classical brown adipose tissue (BAT) and inducible beige adipocytes within white adipose tissue (WAT). Healthy BAT functions to dissipate chemical energy as heat, a process mediated by the mitochondrial uncoupling protein 1 (UCP1). Aging profoundly suppresses sympathetic nerve activity and the expression of UCP1, leading to a precipitous decline in non-shivering thermogenesis and a corresponding reduction in overall organismal energy expenditure 312. Recent transcriptomic studies suggest that the age-related upregulation of the transcription factor forkhead box protein A3 (FOXA3) may be a primary driver behind the loss of BAT activity 3. The accumulation of oxidative stress, diminished thermogenesis, and lipid peroxidation end-products ultimately accelerates the transition of mature adipocytes into a state of irreversible cellular senescence.
Altered Lipid Droplet Dynamics and Adipogenic Exhaustion
Healthy adipose tissue expansion relies on the continuous recruitment and differentiation of mesenchymal progenitor cells in the stromal vascular fraction (SVF). With age, this progenitor pool becomes functionally exhausted and senescent. Advanced scRNA-seq mapping of murine and human white adipose tissue has delineated the developmental trajectory of adipocytes into distinct functional subpopulations, categorizing them from nascent, highly plastic cells (Adipocyte1) to fully mature, highly secretory cells (Adipocyte2). Aging profoundly skews this population dynamic, resulting in an accumulation of immature Adipocyte1 cells and a sharp decline in functional Adipocyte2 cells 8. This developmental arrest reflects an overarching impairment in lipid anabolism and synthetic metabolic capacity 8.
Without the ability to generate new adipocytes, existing cells undergo pathological hypertrophy. These hypertrophic adipocytes struggle to regulate their massive, unilocular lipid droplets. The altered surface-to-volume ratio of the lipid droplet impairs the docking of critical lipolytic enzymes, resulting in defective basal and catecholamine-stimulated lipolysis 413. Furthermore, the sheer physical size of these cells outstrips their local capillary supply, inducing a state of chronic hypoxia that initiates a cascade of fibrotic and inflammatory signaling.
Extracellular Matrix Remodeling and Fibrosis
Perhaps the most mechanically restrictive hallmark of aged and obese adipose tissue is the pathological remodeling of the extracellular matrix (ECM). The ECM provides crucial structural scaffolding, but healthy adipose tissue requires a highly flexible matrix to accommodate dynamic, daily fluctuations in adipocyte volume. In the aged and obese state, localized hypoxia - driven by adipocyte hypertrophy outpacing angiogenesis - stimulates hypoxia-inducible factor 1-alpha (HIF1α) and transforming growth factor-beta (TGF-β) signaling pathways 91415. These pathways trigger the overproduction of ECM components, fundamentally restricting tissue plasticity and instigating adipose tissue fibrosis.
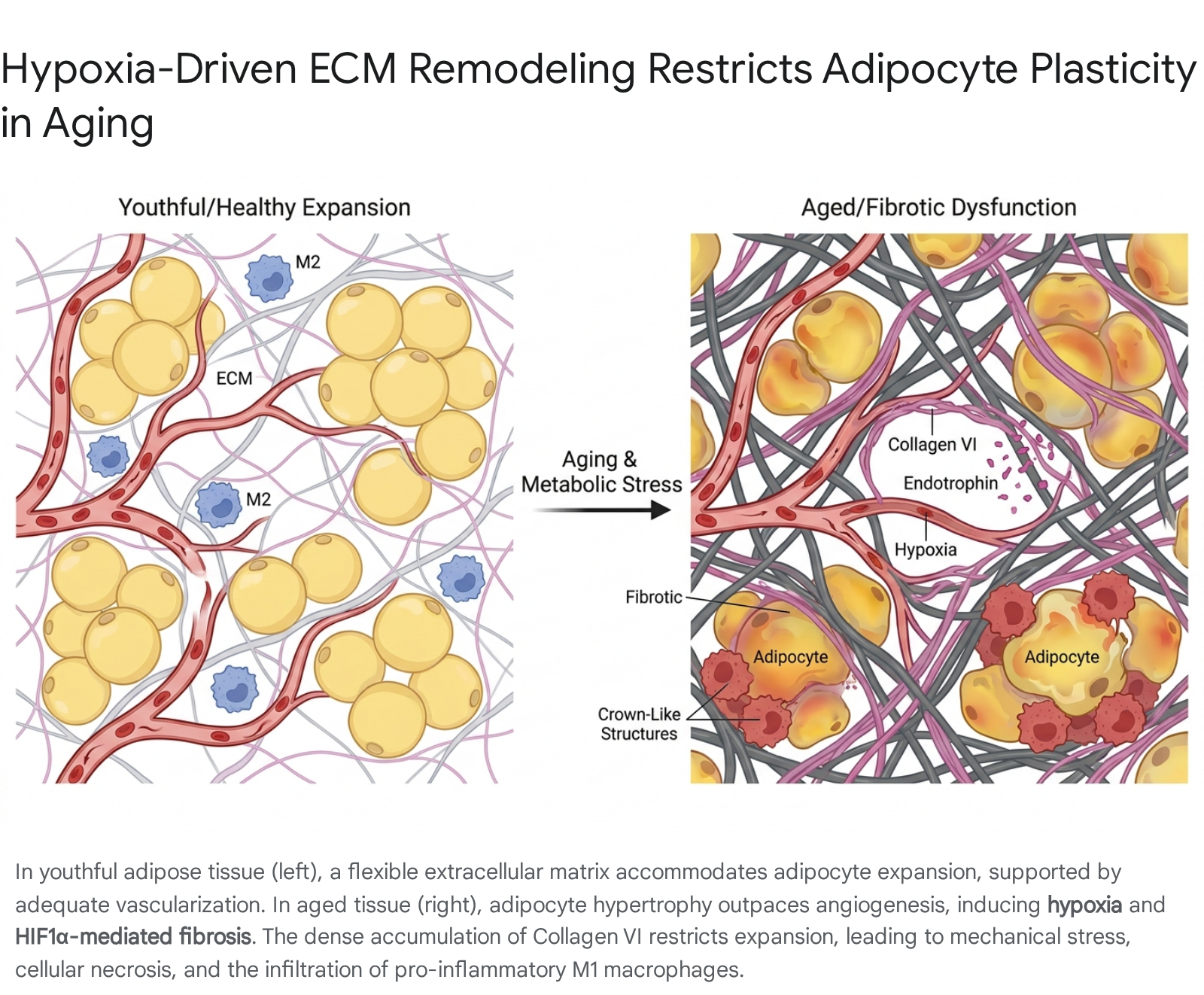
The composition of the ECM shifts dramatically during this fibrotic remodeling. There is a general upregulation of fibrillar and non-fibrillar collagens, notably Collagens I, III, V, and VI 141516. The regulation of these collagens is highly specific to the aging and metabolic context. For example, while the expression of classical fibrillar collagen COL1A1 may actually be downregulated or fragmented in specific aging murine models, there is a massive pericellular accumulation of COL5A1 and COL6A3 in dysfunctional human adipose tissue. Interestingly, clinical data from bariatric surgery cohorts demonstrate that elevated preoperative expression of SAT-derived COL5A1 and COL6A3 strongly predicts insufficient weight loss post-surgery, highlighting how physical matrix rigidity mechanically impedes future lipolysis and adipocyte shrinkage 17.
Type VI Collagen (COL6A1), which is uniquely enriched in adipose tissue, plays an exceptionally deleterious role. The proteolytic cleavage of the α3 chain of Collagen VI produces endotrophin, a potent pro-fibrotic and pro-inflammatory signaling molecule 1417. Elevated endotrophin actively recruits macrophages, triggers local tissue necrosis, and profoundly exacerbates systemic insulin resistance. Conversely, murine models with a genetic deletion of Collagen VI exhibit uninhibited, stress-free expansion of individual adipocytes, paradoxically leading to substantial improvements in whole-body energy homeostasis despite massive obesity 91819. Alongside collagens, other ECM modifiers such as Secreted Protein Acidic and Rich in Cysteine (SPARC), matrix metalloproteinases (MMPs), and tissue inhibitors of metalloproteinases (TIMPs) become highly dysregulated, further cementing the fibrotic architecture 91516.
The rigidification of the pericellular matrix limits cellular plasticity, physically strangling mature adipocytes during periods of energy flux. The resulting mechanotransduction stress eventually causes adipocyte rupture and death. The mechanical death of these cells acts as an intense chemotactic signal, initiating a massive infiltration of immune cells and cementing the transition from localized tissue stress to systemic chronic inflammation.
Immunometabolic Remodeling: Inflammaging and the SASP
Adipose tissue is a premier immunological organ. The stromal vascular fraction contains a vast array of innate and adaptive immune cells that actively maintain tissue homeostasis, buffer lipids, and regulate angiogenesis. Aging, however, provokes a severe dysregulation of this immune landscape, systematically favoring a highly pro-inflammatory environment.
Macrophage Polarization and Adaptive Lymphocyte Shifts
In healthy, youthful adipose tissue, resident adipose tissue macrophages (ATMs) predominantly exhibit an alternatively activated, anti-inflammatory (M2) phenotype. These M2 macrophages secrete interleukin-10 (IL-10), conduct efferocytosis (the clearance of dead cells), regulate local iron flux, and facilitate tissue repair 67. During aging, the total number of macrophages may remain relatively stable, but there is a pronounced phenotypic shift. Driven by hypoxia, circulating free fatty acids, and factors like inositol-requiring enzyme 1α (IRE1α), ATMs transition toward a classically activated, pro-inflammatory (M1) state, alongside the emergence of double-negative (CD206- CD11c-) macrophage subsets 672021. These aggressive M1 macrophages aggregate around necrotic adipocytes to form hallmark "crown-like structures," releasing substantial quantities of tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interleukin-1 beta (IL-1β) directly into the systemic circulation 67.
The adaptive immune system is similarly degraded. The visceral adipose tissue of aged individuals displays a massive accumulation of inflammatory T cells, including a specific expansion of CD8+ cytotoxic T cells and senescent T cells. These senescent T cells exhibit a distinct CD28- CD57+ phenotype, marked by elevated p21 and γH2AX expression, and secrete high levels of interferon-gamma (IFN-γ) 2022. Paradoxically, aging also induces a significant expansion of regulatory T cells (Tregs) within visceral fat, a localized compensatory mechanism that attempts, but ultimately fails, to suppress the overwhelming inflammatory cascade 621. Furthermore, a distinct and highly pathogenic subset known as Age-associated B cells (ABCs), characterized by a CD11c+ CD21- T-bet+ profile, rapidly accumulates in aged visceral fat. These ABCs drive localized autoantibody production and exacerbate insulin resistance 20.
Adipokine Dysregulation and the Adiponectin-Leptin Ratio
The endocrine output of adipose tissue - its adipokine profile - shifts dramatically with age, reflecting the underlying cellular distress. Adiponectin, an insulin-sensitizing, anti-atherogenic, and anti-inflammatory hormone, typically decreases in functionality and concentration in obese and aged populations 12329. While some studies note paradoxically high adiponectin levels in centenarians, longitudinal increases in adiponectin in adults over 65 are often a compensatory response to severe underlying inflammation and are associated with higher mortality 23.
Conversely, leptin, a pro-inflammatory adipokine primarily responsible for signaling satiety to the hypothalamus, increases proportionally with expanding fat mass. However, aging and obesity induce a state of profound central leptin resistance, nullifying its metabolic benefits regarding appetite suppression while its pro-inflammatory peripheral effects on the vasculature persist 12329. The Adiponectin:Leptin (AL) ratio has emerged as a highly sensitive, clinically relevant biomarker for adipose tissue dysfunction. A low AL ratio strongly captures the transition to an inflamed, fibrotic adipose state and inversely correlates with severe cardiometabolic disease risk, offering greater predictive power than either adipokine measured independently 124. Alongside these core hormones, aged adipose tissue also alters the secretion of fibroblast growth factor 21 (FGF21), growth differentiation factor 15 (GDF15), and retinol-binding protein 4 (RBP4), further compounding systemic metabolic dysregulation 125.
The Senescence-Associated Secretory Phenotype and Exosomal Communication
Compounding this adipokine dysregulation is the accumulation of senescent preadipocytes and mature adipocytes. These cells adopt a Senescence-Associated Secretory Phenotype (SASP), secreting a toxic cocktail of inflammatory cytokines, chemokines, and extracellular matrix proteases 262728. Crucially, senescent adipose tissue also alters its output of small extracellular vesicles. Adipose tissue functions as a major source of circulating exosomal microRNAs (miRNAs), which are sorted via specific sequence motifs and RNA-binding proteins. Once released, these exosomal miRNAs act as remote signaling payloads, actively regulating gene expression in distant organs such as the liver and skeletal muscle 28.
This advanced mechanism of inter-organ communication positions adipose tissue not merely as a victim of organismal aging, but as an active, instigating driver. By broadcasting SASP factors and pathogenic exosomal miRNAs into the systemic circulation, senescent adipose tissue acts as an "aging metabolic amplifier," transmitting local cellular senescence into systemic, multi-organ functional decline 2627.
Table 1: Contrasting Young vs. Aged Adipose Tissue Profiles
The transition from youthful metabolic flexibility to aged metabolic dysfunction encompasses coordinated, highly predictable changes across multiple cellular, structural, and systemic domains.
| Biological Feature | Youthful / Healthy Adipose Tissue | Aged / Dysfunctional Adipose Tissue |
|---|---|---|
| Depot Distribution | High functional Subcutaneous Adipose Tissue (SAT); low Visceral Adipose Tissue (VAT). 234 | Progressive loss of SAT capacity; ectopic expansion of VAT and lipid spillover into liver/muscle. 1411 |
| Cellular Morphology | Hyperplastic expansion (adequate recruitment of precursor cells); flexible lipid droplet dynamics. 2915 | Extreme adipocyte hypertrophy; mechanical stress; impaired lipogenesis and basal lipolysis. 4133529 |
| Mitochondrial Function | Efficient oxidative phosphorylation, high mitochondrial dynamics, active UCP1-mediated thermogenesis. 1227 | Mitochondrial DNA damage, excessive ROS production, reduced UCP1 expression, impaired BAT activity. 45 |
| Extracellular Matrix (ECM) | Flexible matrix permitting tissue expansion; balanced MMP and TIMP activity. 915 | Rigid, hypoxic fibrosis; dense accumulation of Collagen VI (endotrophin) and COL5A1; restricted plasticity. 141719 |
| Immune Landscape | Predominance of anti-inflammatory M2 macrophages; stable T-cell homeostasis. 6720 | Shift to pro-inflammatory M1 macrophages; accumulation of Age-associated B cells (ABCs) and senescent T cells. 62022 |
| Endocrine / Secretory | High functional Adiponectin; optimal Leptin sensitivity; healthy exosomal miRNA signaling. 12325 | High Leptin (with leptin resistance); low Adiponectin (low AL ratio); aggressive SASP output amplifying systemic aging. 12427 |
Genetic Determinants and Ethnic Disparities in Adipose Aging
The physiological decline of adipose tissue does not occur uniformly across the human population; it is profoundly and heavily influenced by genetic architecture, epigenetic modifications, and ethnic heritage. An accurate understanding of metabolic risk trajectories must integrate these diverse biological backgrounds.
Master Regulatory Genes and Epigenetic Clocks: The Role of KLF14
Genome-wide association studies (GWAS) utilizing biobanks of unprecedented scale have identified specific genetic loci that govern adipose tissue distribution and metabolic resilience. A premier example is the Krüppel-like factor 14 (KLF14) gene. KLF14 is a maternally-expressed transcription factor that acts as a master trans-regulator of adipose tissue gene expression 3031. Variants near the KLF14 locus (such as rs4731702) are robustly associated with Type 2 Diabetes risk, altered HDL-cholesterol levels, and metabolic syndrome 303240.
Crucially, the metabolic impact of KLF14 is highly sex-specific. In female risk-allele carriers, reduced KLF14 expression leads to a distinct, pathological shift in fat distribution from metabolically protective subcutaneous (gynoid) stores to highly pathogenic visceral (abdominal) depots 10. Fresh adipose tissue explants from female KLF14 risk-allele carriers exhibit a staggering 44% reduction in lipogenesis, fundamentally compromising the tissue's ability to safely store systemic lipids 10. Furthermore, KLF14 acts through downstream targets such as SLC7A10, a solute carrier gene whose downregulation correlates strongly with severe insulin resistance 30. Over time, epigenetic profiling reveals that the KLF14 promoter undergoes age-dependent hypermethylation, suppressing its expression in older and obese individuals. This directly links chronological aging, epigenetic silencing, and accelerating metabolic disease risk 313240. Similar genetic correlations spanning childhood to adulthood have identified variants in FTO and ADCY3 that dictate distinct BMI trajectories, proving that early-life genetic architecture directly influences late-life cardiometabolic disease 3334.
Global Cohorts and Ethnic Adiposity Thresholds
The transition from metabolically healthy to metabolically unhealthy phenotypes differs drastically across ethnic lines, necessitating a departure from traditional, universal Body Mass Index (BMI) guidelines. Substantial evidence from global clinical reviews of the mid-2020s indicates that populations of South Asian, Chinese, Black, and Latin American descent accumulate pathogenic visceral adipose tissue at significantly lower total body weights compared to White cohorts 35363738.
To equate the Type 2 Diabetes incidence rate observed in White adults at a standard obesity BMI of 30.0 kg/m2, equivalent BMI cut-offs must be adjusted drastically downward for non-White populations. In Black women, the equivalent risk occurs at a BMI of 25.9 kg/m2. In South Asian women, the risk threshold drops to 23.3 kg/m2, and in Native Chinese women to an astonishingly low 22.7 kg/m2 3637.
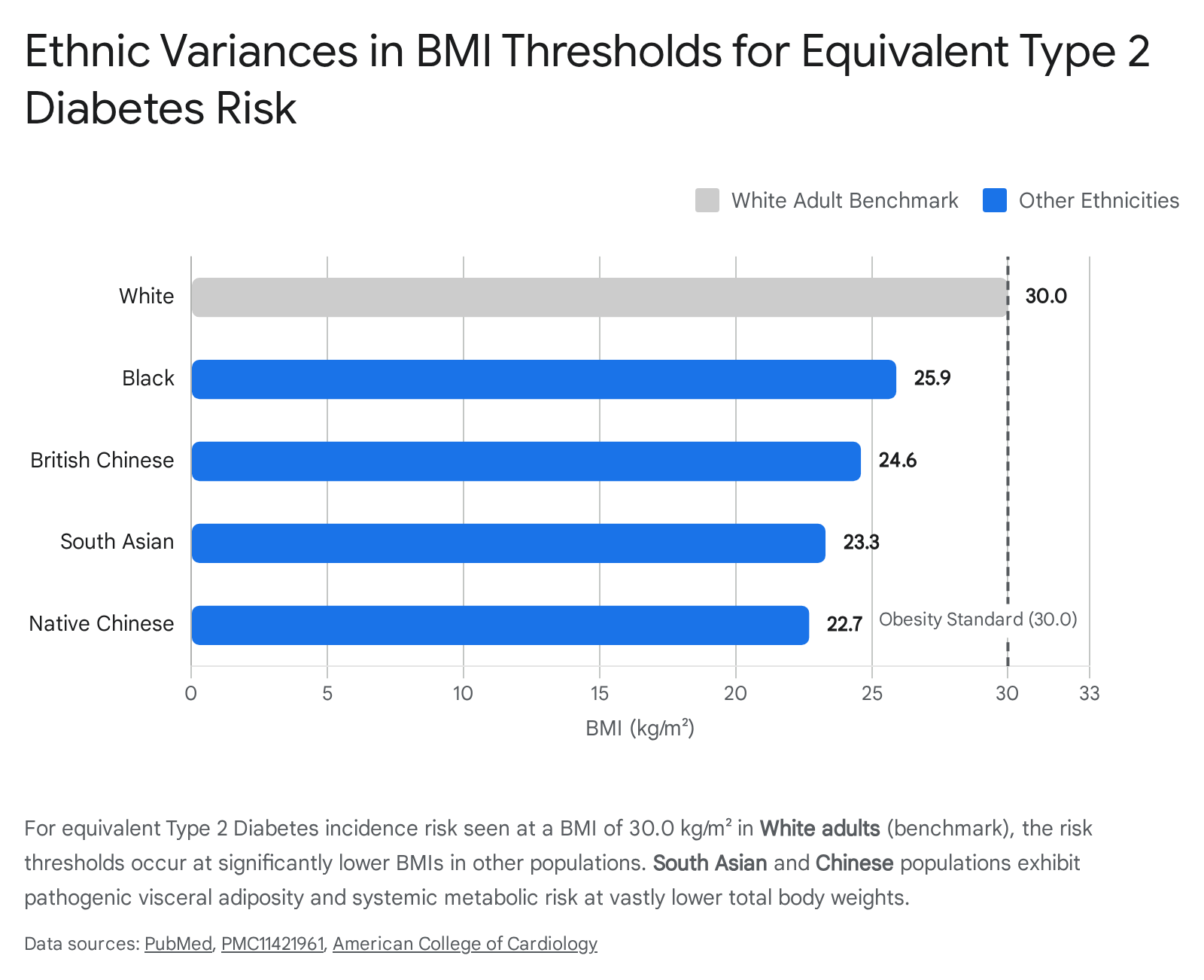
South Asian populations generally possess a smaller skeletal frame but exhibit a much higher propensity for abdominal visceral adiposity - often described as the "thin outside, fat inside" phenotype. This localized visceral fat depot contributes massive quantities of free fatty acids directly into the portal vein vasculature, inducing rapid hepatic steatosis and accelerating systemic insulin resistance long before clinical obesity is diagnosed 3637.
This stark disparity extends to multimorbidity trajectories. Longitudinal data from global aging cohorts, including the English Longitudinal Study of Ageing (ELSA), the Health and Retirement Study (HRS), and the China Health and Retirement Longitudinal Study (CHARLS), reveal striking patterns. Individuals characterized as Metabolically Unhealthy Normal Weight (MUNW) - a phenotype highly prevalent in Asian and minority populations - exhibit aggressive multimorbidity trajectories. In these cohorts, MUNW individuals face a staggering 33.9% probability of entering high-risk trajectory patterns characterized by the rapid co-occurrence of multiple chronic diseases, a risk profile nearly identical to those with classical, visible clinical obesity (Metabolically Unhealthy Overweight/Obesity, MUOO) 3948. Furthermore, research in Latin American cohorts highlights severe regional heterogeneity and sexual dimorphism in abdominal obesity; women consistently display significantly higher prevalence rates under International Diabetes Federation (IDF) criteria than men (74.3% vs. 46.8%), underscoring the urgent need for population- and sex-specific diagnostic thresholds globally 3840.
Therapeutic Interventions: Clinical Translation, Limitations, and Debates
The identification of adipose tissue as a primary pacemaker for systemic aging has catalyzed an explosive era of pharmacological development aimed at rescuing its functionality. While the preclinical rationale for these therapeutics is exceptionally strong, their translation into clinical practice in the mid-2020s is fraught with intense debate, specific biological limitations, and the risk of unintended systemic consequences.
Senolytics: Targeting the Root of Inflammaging
Senolytics represent a novel class of small-molecule therapeutics designed to induce targeted apoptosis in senescent cells by transiently disabling their upregulated pro-survival, anti-apoptotic pathways (SCAPs). The most clinically validated regimen involves the intermittent, "hit-and-run" co-administration of the repurposed chemotherapeutic tyrosine kinase inhibitor Dasatinib (D) and the plant-derived flavonoid Quercetin (Q). Quercetin operates as a strong prooxidant, amplifying oxidative damage particularly in senescent cells that hoard high levels of transition metals like copper and iron, while Dasatinib inhibits Src tyrosine kinase pathways 84142.
Phase 1 and early Phase 2 feasibility trials have successfully provided critical human proof-of-biology. A brief, pulsed oral course of D+Q (typically 100mg Dasatinib and 1250mg Quercetin for two consecutive days every few weeks) in patients with diabetic kidney disease successfully reduced the burden of p16- and p21-expressing senescent cells within adipose tissue and lowered circulating SASP factors 11 days post-treatment 52535455. Similar pilot studies in idiopathic pulmonary fibrosis and early symptomatic Alzheimer's disease have demonstrated favorable tolerability and CNS penetration, alongside reductions in local senescence markers 8105255.
Despite these milestones, the broad clinical translation of senolytics remains deeply debated. The 2026 senescence-targeting landscape indicates that while next-generation molecules - such as the BCL-2/BCL-xL inhibitor Navitoclax (ABT-263) and highly potent Fisetin analogs - demonstrate exceptional efficacy in vitro, a significant fraction of senescent cells rapidly develop resistance mechanisms tied to mitochondrial quality-control 104154. Furthermore, systemic clearance of senescent cells raises profound concerns regarding off-target toxicity. Senescent cells play essential roles in acute wound healing and embryogenesis; their indiscriminate systemic removal could interfere with tissue repair and cause unpredictable long-term epigenetic impacts 4143. The failure of UBX0101, a highly anticipated senolytic, in a rigorous trial for knee osteoarthritis further demonstrated that simply clearing senescent cells does not automatically translate into functional tissue regeneration if the underlying mechanical damage persists 10. Consequently, the most viable near-term clinical applications for senolytics are highly localized interventions - such as Unity Biotechnology's UBX1325 injected directly into the eye for diabetic macular edema - rather than broad, systemic anti-aging protocols 105457.
Incretin Receptor Agonists: The Sarcopenia Conundrum
Glucagon-like peptide-1 (GLP-1) and dual GLP-1/GIP receptor agonists (e.g., Semaglutide, Tirzepatide, Cagrilintide/Semaglutide combos) have revolutionized the treatment of obesity and Type 2 Diabetes by profoundly reducing central appetite and restoring peripheral metabolic homeostasis. Clinical outcomes from massive late-stage trials (SELECT, SURMOUNT) demonstrate unprecedented 15 - 25% body weight reductions. These dramatic reductions in visceral adipose mass are accompanied by significant decreases in major adverse cardiovascular events (MACE), resolutions of liver steatosis, and profound reductions in systemic inflammation 4445606162. In older populations, recent real-world Medicare data suggest that these therapies may actually slow the clinical progression of overall frailty compared to older diabetes medications like DPP-4 inhibitors 6346. Furthermore, emerging data points to a potential reduction in obesity-related cancers, though ongoing trials aim to clarify this association 47.
However, the rapid and massive weight loss induced by these incretin mimetics has ignited a fierce clinical debate regarding the risk of severe sarcopenia in the elderly. Fast weight reduction via extreme caloric deficit frequently results in a disproportionate loss of lean body mass, including critical skeletal muscle 966. For older adults, skeletal muscle is not merely a locomotive organ; it is a vital sink for glucose disposal, a crucial metabolic reserve, and the primary defense against falls and loss of independence. If GLP-1 therapy induces significant lean mass degradation due to severely suppressed protein intake and general fatigue, the cardiometabolic benefits of losing visceral fat may be entirely offset by an increased risk of physical disability and frailty 94766. Consequently, clinical guidelines increasingly mandate that GLP-1 prescribing in geriatric populations be tightly coupled with structured resistance training and aggressive, high-protein nutritional support to mitigate muscle wasting.
Overcoming Anabolic Resistance: Dietary Restriction, Rapamycin, and Metformin
Dietary restriction (DR) has long been the gold standard for extending lifespan and delaying adipose tissue senescence in preclinical models. By reducing the metabolic burden on adipose tissue, DR maintains mitochondrial quality and suppresses adiposity 127. However, imposing strict DR on elderly human populations is clinically precarious. Aging induces profound "anabolic resistance" - a state where the muscle protein synthesis (MPS) machinery becomes blunted, requiring significantly higher thresholds of dietary amino acids (particularly leucine) and mechanical stimuli to activate 114869. To overcome this resistance, guidelines from the PROT-AGE study group recommend elevated protein intakes of 1.0 to 1.2 g/kg/day for healthy older adults, rising to 1.5 g/kg/day for those with chronic illness, distributed optimally across meals 1169. Restrictive diets risk precipitating malnutrition and accelerating sarcopenic decline, negating the metabolic benefits gained by reducing fat mass 1149.
To circumvent the physical risks of strict DR, pharmacological nutrient-sensing modulators that mimic fasting are under intense evaluation: * Rapamycin: As an mTOR inhibitor, rapamycin mimics the biochemical effects of caloric restriction by rebalancing nutrient sensing and enhancing autophagy. The highly anticipated 2025 PEARL (Participatory Evaluation of Aging with Rapamycin for Longevity) trial evaluated low-dose, intermittent compounded rapamycin in healthy older adults over 48 weeks. Strikingly, while systemic adverse events were minimal, women receiving 10 mg weekly demonstrated significant reductions in visceral adipose tissue alongside unexpected, significant improvements in lean muscle mass and self-reported pain 7150515275. This paradigm-shifting result suggests that highly specific, intermittent mTOR inhibition may actually mitigate age-related anabolic resistance rather than exacerbate it, though issues regarding the low bioavailability of compounded formulations require refinement in future trials 715175. * Metformin: Extensively used for decades to treat diabetes, metformin targets fundamental aging pathways by activating AMPK, inhibiting the mitochondrial respiratory chain, and improving mitochondrial quality control via mitophagy 5377. A surprising mechanism discovered recently involves the gut microbiome; metformin dramatically increases the abundance of Akkermansia muciniphila, a bacterium that strengthens the gut lining and lowers systemic inflammation 77. The landmark Targeting Aging with Metformin (TAME) trial aims to establish whether metformin can delay a composite of age-related morbidities (cardiovascular disease, dementia, cancer, mortality) in 3,000 non-diabetic older adults, potentially earning the FDA's first-ever indication for treating "aging" 7854555657. However, debates persist regarding its universal application; emerging 2026 clinical data suggest that in younger, highly physically active individuals, metformin may blunt the necessary mitochondrial stress signals required for optimal exercise adaptation and muscle hypertrophy, suggesting the drug may need to be cycled based on patient age and activity profiles 7778.
Table 2: Interventions Targeting Adipose Aging
The modern therapeutic landscape spans multiple mechanisms of action, each possessing unique levels of clinical validation and distinct physiological risks that must be carefully managed.
| Therapeutic Modality | Primary Agents | Mechanism of Action | Evidence Level / Current Status (2025-2026) | Key Limitations & Debates |
|---|---|---|---|---|
| Senolytics | Dasatinib + Quercetin (D+Q); Fisetin; Navitoclax (ABT-263) | Disables pro-survival pathways (SCAPs) in senescent cells; clears SASP-secreting adipocytes. 84254 | Phase I/II Feasibility. Proven reduction of senescent cells in human adipose tissue (DKD and Alzheimer's trials). 8525354 | Off-target toxicity; cells develop resistance via mitochondrial adaptations; uncertainty regarding long-term systemic use; failed in OA (UBX0101). 104183 |
| Incretin Agonists | Semaglutide (GLP-1); Tirzepatide (GLP-1/GIP); CagriSema | Profound central appetite suppression; reduces visceral metabolic load; attenuates inflammation. 606162 | Approved & Widespread. Strong cardiovascular outcome data (SELECT, SURMOUNT). Shows real-world slowing of frailty. 60616346 | High risk of Sarcopenia in the elderly due to rapid loss of lean muscle mass; absolute requirement for high protein intake and resistance training. 966 |
| mTOR Inhibitors | Rapamycin (Rapalogs) | Mimics caloric restriction; modulates nutrient sensing; suppresses hyperactive growth signals. 5771 | PEARL Trial (2025) demonstrates safety; preserves lean mass and reduces VAT specifically in aging women. 715052 | Bioavailability issues with compounded versions; optimal dosing frequency (pulsed vs. daily) to avoid immunosuppression remains unclear. 715175 |
| AMPK Activators | Metformin | Enhances mitochondrial mitophagy; improves peripheral insulin sensitivity; beneficially alters gut microbiome (Akkermansia). 5377 | TAME Trial underway. Supported by extensive retrospective epidemiological data showing reduced mortality in diabetics. 77545684 | May blunt beneficial mitochondrial adaptations to physical exercise in younger/active populations; common gastrointestinal distress. 7778 |
Conclusion
The biological reality of human aging is inextricably linked to the structural and functional deterioration of the adipose organ. Far from being a passive storage depot, adipose tissue operates as a master endocrine regulator. As chronological age advances, this tissue transitions from a highly flexible, protective compartment into a fibrotic, hypoxic, and senescent mass that actively propagates systemic decline. This pathological transition - driven fundamentally by mitochondrial exhaustion, restricted extracellular matrix expandability, and the aggressive, wide-ranging secretion of pro-inflammatory SASP factors and exosomal miRNAs - cements adipose tissue as a central pacemaker in the chronology of human aging and metabolic disease.
Crucially, recognizing that physiological decline is highly heterogeneous, modern precision medicine must account for profound ethnic and genetic variances. Diverse genetic architectures, governed by master trans-regulators like KLF14, combined with distinct ancestral propensities for rapid visceral fat accumulation, demonstrate unequivocally that classical anthropometric benchmarks are insufficient for global risk assessment. Populations of South Asian, Chinese, and Latin American descent face severe metabolic risks at body weights traditionally deemed "healthy," necessitating a paradigm shift toward individualized metabolic profiling.
Consequently, the therapeutic landscape is evolving at an unprecedented pace. While the massive weight reductions achieved by modern incretin therapies and the targeted cellular clearance promised by the first generation of senolytics represent monumental leaps in metabolic geroscience, their clinical application requires extreme nuance. Mitigating the profound risks of anabolic resistance, off-target toxicity, and age-related sarcopenia remains the ultimate barrier to widespread implementation. Future geroprotective interventions must not merely aim to reduce total adiposity, but must fundamentally repair the cellular microenvironment of adipose tissue - resolving fibrosis, restoring mitochondrial dynamics, and preserving its vital endocrine function - thereby safely extending human healthspan.