The 2023 hallmarks of aging framework and interactions
Introduction to Biological Aging Models
Biological aging is characterized by a progressive, time-dependent loss of physiological integrity, leading to impaired function and an exponential increase in vulnerability to death. It is the primary risk factor for the most prevalent human pathologies, including cardiovascular diseases, neurodegeneration, metabolic syndromes, and cancer 123. For decades, gerontological research lacked a unified paradigm to categorize the diverse cellular and molecular deteriorations observed across species. In 2013, López-Otín and colleagues established a foundational framework by defining nine molecular and cellular "hallmarks of aging," establishing a conceptual scaffold analogous to the recognized hallmarks of cancer 245.
A decade of subsequent research and therapeutic advancements necessitated an evolution of this framework. In 2023, the model was expanded to encompass twelve distinct but highly interconnected hallmarks 567. To qualify as a hallmark within this paradigm, a biological process must satisfy three strict criteria: it must manifest during normal physiological aging, its experimental aggravation must accelerate the aging phenotype, and its experimental alleviation must decelerate, halt, or reverse aging, thereby extending healthy lifespan (healthspan) 468.
The expanded 2023 framework organizes the twelve hallmarks into a functional, three-tier hierarchy: primary hallmarks (the root causes of cellular damage), antagonistic hallmarks (compensatory responses that become deleterious when chronically activated), and integrative hallmarks (the systemic culmination of homeostatic failure) 6. This structural categorization provides a precise map for geroscience, shifting the focus from treating isolated age-related diseases toward targeting the fundamental biological drivers of senescence 89.
Primary Hallmarks of Aging
The primary hallmarks are unequivocally negative drivers of aging. They initiate the process through the progressive accumulation of molecular and cellular damage over an organism's lifespan. These hallmarks represent the breakdown of essential maintenance systems that preserve the fidelity of DNA, epigenetic regulation, and protein homeostasis 2610.
Genomic Instability
The integrity of the genome is continuously challenged by extrinsic stressors, such as ultraviolet radiation and chemical mutagens, and intrinsic threats, including DNA replication errors and reactive oxygen species (ROS) produced by cellular metabolism 1112. It is estimated that mammalian DNA sustains up to one million damaging events per cell per day 11. While cells possess robust DNA repair mechanisms - including homologous recombination and non-homologous end joining - these systems are imperfect and decline in efficiency with age 1114.
The resulting genomic instability manifests as point mutations, translocations, chromosomal aneuploidies, and the accumulation of double-strand breaks 5. This mutational burden drives cellular senescence, impairs tissue function, and severely increases oncogenic risk 1114. Comparative genomics studies analyzing multiple mammalian species indicate that a species-specific rate of somatic mutation is inversely correlated with maximum lifespan, underscoring genomic instability as a fundamental molecular clock of aging 5. Deficiencies in DNA repair pathways also directly cause premature aging conditions, such as Werner syndrome and Hutchinson-Gilford progeria syndrome 1314.
Telomere Attrition
Telomere attrition is a specialized form of genomic instability. Telomeres are repetitive nucleotide sequences bound by the shelterin protein complex, capping the ends of linear chromosomes to protect them from degradation and from being falsely recognized as DNA double-strand breaks 111417. Due to the "end-replication problem," DNA polymerase cannot fully replicate the lagging strand during cell division, causing telomeres to shorten with each cycle 1715.
When telomeres reach a critical length, the shelterin complex is disrupted, triggering a permanent DNA damage response that halts the cell cycle - a state known as replicative senescence 141516. While the enzyme telomerase can restore telomere length, its expression in adult somatic tissues is highly restricted to the germline and specific stem cell compartments 1115. This repression acts as a crucial evolutionary safeguard against unchecked cellular proliferation and tumor growth, representing a primary biological trade-off between cancer prevention and tissue aging 1115.
Epigenetic Alterations
Aging is accompanied by profound changes to the epigenome, the secondary layer of information that regulates gene expression without altering the underlying DNA sequence. Epigenetic alterations include DNA methylation drift, changes in histone modifications, and broad-scale chromatin remodeling 111217.
Throughout an organism's lifespan, the precisely tuned epigenetic landscape of youth begins to lose its fidelity. Promoters of essential genes may become hypermethylated and silenced, while normally repressed genomic regions, such as retrotransposons and heterochromatin, may become hypomethylated and transcriptionally active 1620. This increase in transcriptional noise compromises cellular identity. For instance, SIRT1 and other chromatin-modifying proteins that normally maintain epigenetic silencing are frequently recruited away from their loci to assist in repairing double-strand DNA breaks. Over time, this repeated relocalization degrades the original epigenetic landscape, contributing to cellular dysfunction and phenotypic aging 217. The predictability of these methylation changes across multiple tissues has enabled the development of "epigenetic clocks," which estimate biological age based on specific DNA methylation signatures 1823.
Loss of Proteostasis
Protein homeostasis, or proteostasis, ensures that proteins are correctly folded, assembled, localized, and ultimately degraded when damaged or no longer needed 111715. The cellular proteostasis network involves molecular chaperones, which assist in proper protein folding, and two primary proteolytic systems: the ubiquitin-proteasome system and autophagy 11.
With advancing age, the fidelity of protein synthesis and the efficiency of quality-control mechanisms decline. This leads to the accumulation of misfolded, oxidized, and damaged proteins that form toxic intracellular aggregates 1215. Loss of proteostasis is highly visible in age-related neurodegenerative conditions, such as Alzheimer's disease, characterized by amyloid-beta and tau tangles, and Parkinson's disease, driven by alpha-synuclein aggregates 1315. The inability to clear these aberrant proteins triggers chronic cellular stress pathways, impairs normal organelle function, and contributes directly to tissue toxicity and cell death 1925.
Disabled Macroautophagy
Initially considered a sub-component of proteostasis in the 2013 framework, disabled macroautophagy was elevated to a distinct primary hallmark in the 2023 update due to its broad physiological impacts beyond simple protein degradation 62627. Macroautophagy is a highly conserved cellular recycling process in which double-membrane vesicles called autophagosomes engulf damaged organelles, toxic protein aggregates, and intracellular pathogens, delivering them to lysosomes for degradation and metabolic recycling 62627.
Autophagy is crucial for maintaining cellular homeostasis. However, autophagic flux declines progressively with age. The mechanistic target of rapamycin (mTOR) pathway is a primary negative regulator of autophagy; age-associated hyperactivation of mTOR chronically suppresses autophagosome formation 132620. The failure to sequester and degrade dysfunctional cellular components, particularly aging mitochondria (a specific form of macroautophagy known as mitophagy), leads to catastrophic accumulations of cellular debris. This disabled clearance accelerates the deterioration of highly post-mitotic cells, such as neurons and cardiomyocytes, disrupting cellular metabolic balance and driving the progression of age-related diseases 131727.
Antagonistic Hallmarks of Aging
Antagonistic hallmarks are distinct from primary hallmarks in their duality. In youth or at low intensities, these processes are beneficial and necessary for survival, mediating adaptive responses to stress or nutrient availability. However, as age progresses and primary damage accumulates, these once-protective mechanisms become chronically hyperactivated or severely dysregulated, effectively transitioning into powerful drivers of the aging process 5614.
Deregulated Nutrient Sensing
The nutrient-sensing network is an archetypal example of antagonistic pleiotropy - biological processes that enhance evolutionary fitness early in life but cause harm in post-reproductive stages 621. Cells rely on intricate signaling pathways to detect the presence or scarcity of nutrients, dictating whether the cell should enter an anabolic state of growth and proliferation or a catabolic state of repair and recycling 17.
Key nodes in this network include the insulin and insulin-like growth factor 1 (IGF-1) signaling pathway, the mTOR kinase, AMP-activated protein kinase (AMPK), and the sirtuin (SIRT) family 21325. In a youthful state, abundant nutrients activate mTOR and IGF-1, promoting tissue growth and development. Conversely, nutrient scarcity activates AMPK and sirtuins, which upregulate stress resistance, DNA repair, and autophagy 17. During aging, this precise regulatory system becomes deregulated. Chronic nutrient abundance and age-related metabolic inflexibility lead to continuous mTOR activation and systemic insulin resistance, suppressing essential autophagic repair processes and promoting abnormal fat storage and cellular exhaustion 1317.
Mitochondrial Dysfunction
Mitochondria are the primary sites of cellular respiration and ATP production. Over time, mitochondrial efficacy declines due to a combination of mitochondrial DNA (mtDNA) mutations, destabilization of electron transport chain complexes, altered mitochondrial fission and fusion dynamics, and impaired mitophagy 21216.
A byproduct of oxidative phosphorylation is the generation of reactive oxygen species (ROS). Historically, the "free radical theory of aging" posited that accumulating ROS directly caused aging through widespread macromolecular damage 21. Modern geroscience has nuanced this view. At low, transient levels, ROS act as critical signaling molecules that trigger compensatory survival pathways - a phenomenon known as mitohormesis 610. However, as mitochondrial dysfunction becomes severe and chronic with age, the resulting high-level ROS production exceeds cellular antioxidant capacity, triggering pervasive oxidative stress, generalized inflammation, and widespread cellular damage 21216.
Cellular Senescence
Cellular senescence is a state of stable and irreversible cell-cycle arrest. It operates initially as a protective mechanism triggered by telomere attrition, DNA double-strand breaks, or oncogene activation to prevent the dangerous proliferation of damaged, potentially malignant cells 141722. Senescence is also essential during embryonic development and for acute wound healing and tissue repair 16.
Despite their arrested replication, senescent cells are metabolically active. They undergo profound chromatin remodeling and develop a Senescence-Associated Secretory Phenotype (SASP) 2324. The SASP involves the continuous local secretion of a complex cocktail of pro-inflammatory cytokines (such as IL-6 and IL-1$\beta$), chemokines, growth factors, and matrix metalloproteinases 2526. In youth, the immune system rapidly clears these aberrant cells. In older organisms, immune clearance falters, leading to the gradual accumulation of senescent cells in tissues. The chronic local and systemic secretion of SASP factors degrades the surrounding extracellular matrix, impairs neighboring healthy cells, limits tissue regenerative capacity, and acts as a primary physiological driver for aging-related chronic inflammation 252324.
Integrative Hallmarks of Aging
Integrative hallmarks represent the systemic culmination of the aging process. They emerge when the accumulated damage from the primary hallmarks and the resulting dysregulation of the antagonistic hallmarks overwhelm the organism's homeostatic compensatory mechanisms. These hallmarks dictate the clinical manifestations of aging, resulting in tissue failure, frailty, and chronic disease 6810.
Stem Cell Exhaustion
Tissue homeostasis depends on the continuous renewal of cells by resident stem cell populations. Over time, the regenerative potential of these stem cell pools diminishes, a process termed stem cell exhaustion 6815. This exhaustion is the direct downstream consequence of multiple upstream hallmarks, including telomere attrition, accumulation of DNA mutations, epigenetic drift, and the hostile, inflammatory microenvironment created by chronic SASP 1015.
As stem cells undergo apoptosis, enter senescence themselves, or lose their pluripotency, the tissue's ability to repair damage and regenerate functional parenchyma is severely compromised. This is clinically observable in the aging of the hematopoietic system (leading to immunosenescence and age-related anemias), the depletion of intestinal crypt stem cells, and the loss of skeletal muscle satellite cells, which directly contributes to age-related muscle wasting, or sarcopenia 132527.
Altered Intercellular Communication
Aging is not merely a cell-autonomous event; it profoundly alters how cells and organ systems coordinate across the body. Altered intercellular communication involves derangements in endocrine, neuroendocrine, and neuronal signaling pathways 825.
This hallmark includes pathological changes to the extracellular matrix (ECM). The ECM becomes increasingly stiff due to advanced glycation end-products (AGEs) and the structural breakdown of collagen networks, mechanically impeding cellular function and tissue elasticity 625. Furthermore, aging alters the composition of blood-borne systemic factors. Heterochronic parabiosis experiments - where the circulatory systems of a young and an old mouse are surgically joined - have demonstrated that factors in old blood can actively induce aging phenotypes in young tissues, while youthful circulating factors can partially rejuvenate aged organs, proving that aging operates as a highly integrated systemic network 517.
Chronic Inflammation (Inflammaging)
Added as a distinct hallmark in 2023, chronic inflammation - often termed "inflammaging" - is a pervasive, low-grade, sterile (non-infectious) inflammatory state that characterizes biological aging 232829. While acute inflammation is vital for host defense and repair, persistent inflammatory signaling damages tissues, accelerates telomere shortening, and disrupts metabolic homeostasis.
Inflammaging represents the systemic aggregation of signals originating from senescent cells via the SASP, the accumulation of misfolded proteins and cytosolic DNA, and the general decline in immune system efficacy, known as immunosenescence 2330. Recent discoveries indicate that macrophages, a key immune cell type, lose their ability to properly utilize calcium signaling in their mitochondria as they age. This energetic failure renders them hyperactive, perpetually releasing inflammatory cytokines even in the absence of pathogens 31. The resulting chronic inflammation acts as a central hub, exacerbating cardiovascular disease, neurodegeneration, and insulin resistance, forming a vicious positive feedback loop with other aging hallmarks 2829.
Microbiome Dysbiosis
The human gastrointestinal tract hosts trillions of microorganisms, engaging in a symbiotic relationship vital for nutrient digestion, immune regulation, and maintenance of the gut epithelial barrier 4032. Microbiome dysbiosis, recognized as the twelfth hallmark in the 2023 update, refers to the age-associated loss of microbial diversity and the structural shift toward pathogenic profiles 64033.
During aging, populations of beneficial, short-chain fatty acid (SCFA)-producing bacteria (such as Bifidobacterium) decline, while populations of pro-inflammatory bacteria (such as Proteobacteria) increase 324334. SCFAs like butyrate are essential for maintaining the tight junctions of the intestinal epithelium and the blood-brain barrier 3243. A reduction in these metabolites compromises gut barrier integrity, leading to intestinal permeability or "leaky gut." This permits bacterial endotoxins, such as lipopolysaccharides (LPS), to translocate into the systemic circulation, directly triggering Toll-like receptors and fueling systemic inflammaging. Dysbiosis is highly implicated in the gut-brain axis, correlating strongly with cognitive decline and neurodegenerative diseases like Alzheimer's and Parkinson's 273343.
Systemic Interconnectivity and Hierarchical Crosstalk
The twelve hallmarks of aging do not operate in isolation; they are bound by formidable interconnectivity 61014. Modulating a single hallmark experimentally inevitably cascades through the biological network, either alleviating or exacerbating multiple other aging processes simultaneously 1435.
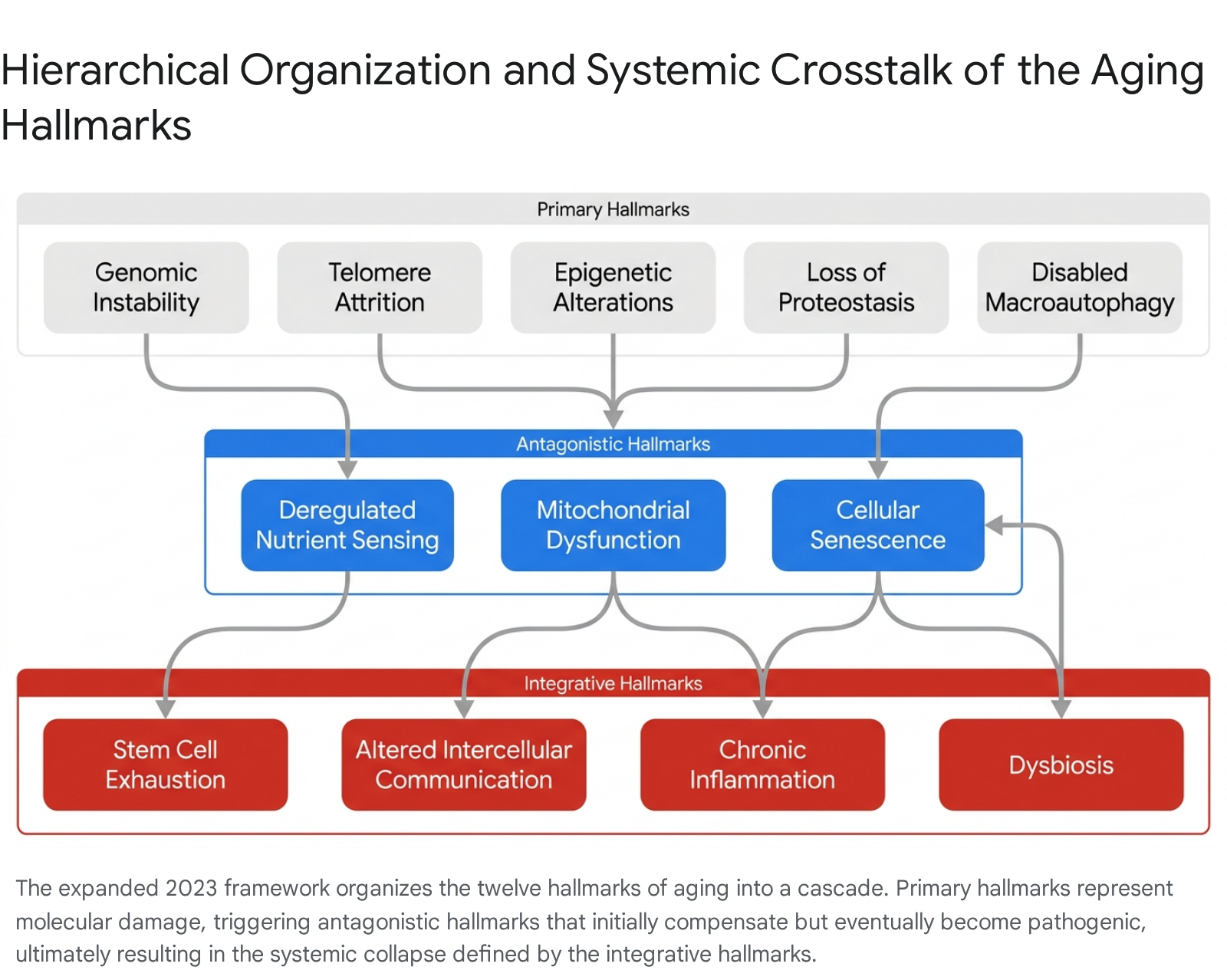
The hierarchical flow of biological aging begins at the molecular level. For instance, genomic instability directly drives epigenetic alterations by necessitating the constant mobilization of chromatin-modifying enzymes away from their regulatory sites to locations of DNA repair 217. Furthermore, unrepaired DNA damage and critically short telomeres explicitly trigger the $p53$ and $p16^{INK4a}$ signaling pathways, forcing cells into protective cellular senescence 222.
Once senescence is established, the SASP releases pro-inflammatory cytokines, acting as a bridge to the integrative hallmarks by driving chronic systemic inflammation. This local inflammatory environment suppresses the regenerative capacity of nearby stem cells, directly resulting in stem cell exhaustion 102527. The 2023 additions to the framework heavily underscore these systemic interactions. For instance, disabled macroautophagy is deeply intertwined with gut dysbiosis and inflammaging 3246. When macroautophagy is disabled by hyperactive nutrient sensing, the gut barrier degrades. Simultaneously, age-related dysbiosis reduces SCFA availability, which further weakens epithelial tight junctions. This synergistic failure allows microbial endotoxins to penetrate the systemic circulation, violently triggering chronic inflammation, which in turn increases oxidative stress, driving further mitochondrial dysfunction and DNA damage 2733.
To conceptualize how the 12 hallmarks interact with emerging therapeutic strategies, the following matrix summarizes the framework's categorization, mechanistic drivers, and the active geroscience interventions targeting them.
| Hallmark Category | Specific Hallmark | Primary Pathological Mechanism | Prominent Therapeutic Interventions |
|---|---|---|---|
| Primary | Genomic Instability | Accumulation of DNA mutations and double-strand breaks | PARP activators, NAD+ precursors (NMN, NR) 1320 |
| Primary | Telomere Attrition | Erosion of chromosome ends triggering cell cycle arrest | Telomerase gene therapy (TERT), TA-65 13 |
| Primary | Epigenetic Alterations | Loss of DNA methylation fidelity and chromatin structure | Partial epigenetic reprogramming (OSK) 1347 |
| Primary | Loss of Proteostasis | Accumulation of toxic misfolded proteins | HSP inducers, Proteasome activators 1340 |
| Primary | Disabled Macroautophagy | Failure of lysosomal degradation of damaged organelles | mTOR inhibitors (Rapamycin), Spermidine 1013 |
| Antagonistic | Deregulated Nutrient Sensing | Hyperactivation of mTOR and IGF-1; decline in AMPK | Metformin, Acarbose, Caloric Restriction 1317 |
| Antagonistic | Mitochondrial Dysfunction | Decline in oxidative phosphorylation and elevated ROS | Mitophagy inducers (Urolithin A), NAD+ boosters 1336 |
| Antagonistic | Cellular Senescence | Accumulation of growth-arrested cells emitting SASP | Senolytics (Dasatinib + Quercetin, Fisetin) 413 |
| Integrative | Stem Cell Exhaustion | Depletion of regenerative tissue capacity | Mesenchymal stem cell therapy, GDF11 13 |
| Integrative | Altered Intercellular Comm. | Scrambled neuroendocrine signaling and stiffened ECM | Exosome therapy, blood factor rejuvenation 1320 |
| Integrative | Chronic Inflammation | Sterile, low-grade systemic immune activation | IL-1/IL-6 inhibitors, Aspirin, Senolytics 1329 |
| Integrative | Dysbiosis | Loss of beneficial microbiota and intestinal barrier failure | Probiotics, Prebiotics, Fecal Microbiota Transplant 4034 |
Theoretical Critiques of the Aging Paradigm
While the hallmarks of aging framework has successfully provided an actionable scaffold for geroscience, it has faced robust critique regarding its explanatory power. Several theoretical frameworks challenge the assumption that aging is simply a consequence of stochastic molecular damage.
The Hyperfunction Theory and Multifactorial Models
Biogerontologists such as David Gems and João Pedro de Magalhães have argued that the hallmarks framework represents a descriptive taxonomy rather than a true scientific paradigm 373839. In their critique, often framed by the "Hoverfly and the Wasp" metaphor (suggesting the hallmarks mimic a paradigm without providing deep mechanistic explanation), they argue the hallmarks catalog the symptoms of aging rather than the root evolutionary causes 3852.
The "damage accumulation" theory - the philosophical foundation of the primary hallmarks - suggests aging is akin to the passive wear and tear of a machine. Conversely, the Hyperfunction Theory, originally championed by Mikhail Blagosklonny and supported by Gems, posits that aging is not a passive decay but an active, deleterious continuation of early-life developmental and reproductive genetic programs 5253. Under this theory, aging is a quasi-program driven by wild-type gene action. The hyperactivation of developmental pathways, such as mTOR, in post-reproductive life leads to cellular hypertrophy and hyperfunction, generating senescent pathologies 5253. Gems proposes a two-stage multifactorial model: an initial stage of environmental and genetic disruption, followed by a second stage where wild-type genetic programs fail to shut off, driving the late-life pathologies recognized as age-related disease 53.
Evolutionary Constraints and Systems Biology
João Pedro de Magalhães emphasizes that the hallmarks framework often neglects the evolutionary and systems-level context of aging. Different species exhibit vastly different lifespans despite sharing the same fundamental biochemistry - a mouse ages in three years while a bowhead whale lives over two centuries 54. By utilizing network and machine learning approaches to analyze species with exceptional longevity, this perspective highlights that aging must be understood as a dynamic, system-level process shaped by evolutionary trade-offs, not merely as a collection of isolated molecular targets 544041. Understanding these cross-species genomic networks provides a more rigorous backbone for distinguishing biologically plausible longevity strategies from speculative interventions 40.
The Information Theory of Aging
Another leading alternative framework is the Information Theory of Aging (ITOA), advanced by David Sinclair and colleagues. While the traditional hallmarks prioritize physical DNA mutations as the core driver, the ITOA posits that aging is fundamentally driven by a loss of epigenetic, rather than genetic, information 205742.
Cells store information in two ways: the digital genome (DNA sequence) and the digital-analog epigenome (methylation and histone modifications that dictate cell identity) 42. Sinclair's team hypothesizes that as cells continually repair DNA double-strand breaks over a lifespan, chromatin modifiers are repeatedly pulled away from their primary regulatory sites. This "relocalization of chromatin modifiers" leaves behind epigenetic noise 171857. Over time, cells experience "ex-differentiation" - they forget their specialized cellular identity, leading to widespread tissue dysfunction 20. Crucially, the ITOA suggests mammals retain a backup copy of youthful epigenetic information. If accessed via epigenetic reprogramming, it implies biological aging is largely reversible, akin to reinstalling corrupted software 1859.
Common Misconceptions in Aging Science
The rapid expansion of the longevity industry has also necessitated the debunking of several pervasive myths. Aging biologist Matt Kaeberlein emphasizes that current technologies, including highly publicized epigenetic clocks, cannot directly measure an individual's precise biological age; rather, they provide estimations based on molecular correlations with mortality risk 2324. Furthermore, a common misconception is that longevity interventions are only effective if initiated early in life. Scientific evidence from animal models using geroprotectants like rapamycin demonstrates that initiating treatments in older subjects can not only delay further functional decline but occasionally reverse it 2360.
Clinical Translation of Gerotherapeutics
The translation of the hallmarks framework from model organisms to human clinical applications is the primary objective of contemporary geroscience 82043. Currently, multiple pharmaceutical interventions are navigating clinical trials, targeting distinct pillars of the aging network to increase healthspan and prevent multimorbidity.
Nutrient-Sensing Modulators: Rapamycin and Metformin
Inhibiting overactive nutrient-sensing pathways is a heavily validated strategy for extending lifespan in animal models. Rapamycin, an FDA-approved immunosuppressant, acts as a direct inhibitor of the mTOR complex, mimicking the biochemical effects of caloric restriction and upregulating macroautophagy 132062.
While chronic, high-dose rapamycin administration carries risks of immunosuppression and insulin resistance, intermittent low-dose regimens show remarkable promise for healthy aging 6344. The Participatory Evaluation of Aging with Rapamycin for Longevity (PEARL) trial - a 48-week, double-blind, randomized controlled study - evaluated intermittent low-dose rapamycin (5 mg and 10 mg weekly) in a normative aging human cohort. The trial reported no significant differences in adverse events compared to placebo 6545. While longitudinal data on lifespan extension remains pending, the trial recorded significant, sex-specific improvements. Women in the 10 mg cohort experienced a 4.5% increase in lean tissue mass and significant reductions in self-reported pain scores, while men in the 10 mg cohort exhibited a 1.4% increase in bone mineral content, suggesting targeted preservation of musculoskeletal healthspan 456768.
Metformin, a first-line type 2 diabetes medication, acts as a senomorphic and an AMPK activator, altering cellular energy metabolism 1346. Large retrospective observational studies have controversially suggested that diabetic patients taking metformin may outlive matched non-diabetic controls 6246. To test its geroprotective effects formally, the NIH-supported Targeting Aging with Metformin (TAME) trial has been designed. TAME will assess whether metformin can delay the composite onset of major age-related diseases - including cancer, cardiovascular disease, and cognitive decline - in a large cohort of older adults 624647.
Senolytics and Senomorphic Agents
Targeting cellular senescence remains one of the most active translational pipelines. Senolytics are agents designed to selectively induce apoptosis in senescent cells by disabling their unique anti-apoptotic survival pathways, while senomorphics aim to suppress the secretion of the SASP without killing the cell 417.
The combination of the tyrosine kinase inhibitor Dasatinib and the flavonoid Quercetin (D+Q) has shown significant efficacy in early clinical studies. In a human trial for diabetic kidney disease, D+Q administration successfully depleted p16- and p21-positive senescent cells and reduced circulating SASP factors, leading to improvements in kidney function 174849. Single-agent senolytics are also progressing. The BCL-xL inhibitor UBX1325 has recently shown statistical and clinical improvements in phase IIa trials for patients with diabetic macular edema, validating the targeted senolytic approach in localized human tissues 49.
Emerging strategies are advancing beyond small molecules to senolytic vaccines. Recent research highlights targeting specific senescent cell surface proteins, such as Glycoprotein nonmetastatic melanoma protein B (GPNMB) or CD153, to stimulate the immune system to clear senescent endothelial and immune cells. This targeted immunoclearance has successfully reduced atherosclerotic plaques and improved metabolic dysfunction in rodent models, pointing to highly specific, next-generation longevity therapeutics 132250.
Partial Epigenetic Reprogramming
The most ambitious therapeutic translation targets the epigenetic hallmark through cellular reprogramming. Utilizing the Yamanaka transcription factors - specifically Oct4, Sox2, and Klf4 (OSK) - researchers have demonstrated the capacity to erase age-related epigenetic marks and reset a cell to a youthful, functional state 575974.
Traditional full reprogramming transforms somatic cells entirely into pluripotent stem cells, erasing their functional identity and posing severe teratoma (tumor) risks 7475. Consequently, longevity researchers employ partial or transient reprogramming. By cycling the activation of the OSK factors, researchers aim to roll back epigenetic aging clocks while allowing the cell to maintain its specialized identity 7476.
Preclinical outcomes have been highly successful. Systemic delivery of AAV-encoded OSK factors in aged mice has extended median remaining lifespan by over 100% and reversed biological age markers in cardiac and hepatic tissues 47. Leading biotech companies are pushing this technology toward the clinic. Life Biosciences is advancing its Partial Epigenetic Reprogramming (PER) platform, utilizing OSK gene therapy to successfully restore visual function in non-human primate models of optic neuropathy. The company anticipates initiating IND-cleared Phase 1 human trials for these ocular indications in early 2026 7577. If translated successfully to humans, partial reprogramming represents a paradigm shift from merely decelerating damage to actively rejuvenating human tissue.
| Therapeutic Category | Lead Candidates | Target Hallmark(s) | Current Clinical Status |
|---|---|---|---|
| mTOR Inhibitors | Rapamycin | Nutrient Sensing, Macroautophagy | Phase II/III (e.g., PEARL Trial showing safety/lean mass gains) 6545 |
| AMPK Activators | Metformin | Nutrient Sensing, Inflammation | Phase III planning (TAME Trial composite disease outcomes) 6246 |
| Senolytics | Dasatinib + Quercetin | Cellular Senescence | Phase I/II (Demonstrated SASP clearance in diabetic kidney disease) 49 |
| Senolytics | UBX1325 (BCL-xL inhibitor) | Cellular Senescence | Phase IIa (Vision improvement in diabetic macular edema) 49 |
| Reprogramming | OSK Gene Therapy | Epigenetic Alterations | Preclinical / IND-clearing (Human trials expected 2026 for optic neuropathy) 7577 |
| NAD+ Boosters | Nicotinamide Riboside (NR) | Mitochondrial Dysfunction | Phase I/II (Demonstrated improvements in exercise performance) 6247 |
Clinical Implementation of Longevity Medicine
The rapid progression of geroscience from bench to bedside requires specialized clinical infrastructure capable of delivering these advanced therapeutics. Recognizing this need, major medical institutions are establishing dedicated clinical centers.
The National University Health System (NUHS) in Singapore launched the world's first Healthy Longevity Clinic within a public hospital setting at Alexandra Hospital 517952. Directed by researchers such as Andrea Maier and Brian Kennedy, the clinic utilizes precision geromedicine to measure an individual's biological age through advanced epigenetic and functional biomarkers specific to the local population 5152. The clinical objective is to translate biological interventions into practice to increase the healthspan of the population by three years over the next decade. Patients at the clinic are evaluated for targeted, evidence-based interventions - ranging from highly personalized exercise regimens and supplements to participation in ongoing clinical trials for repurposed geroprotective drugs 7952. This model represents the future operationalization of the hallmarks framework, shifting healthcare systems from reactive disease management to proactive biological age mitigation.